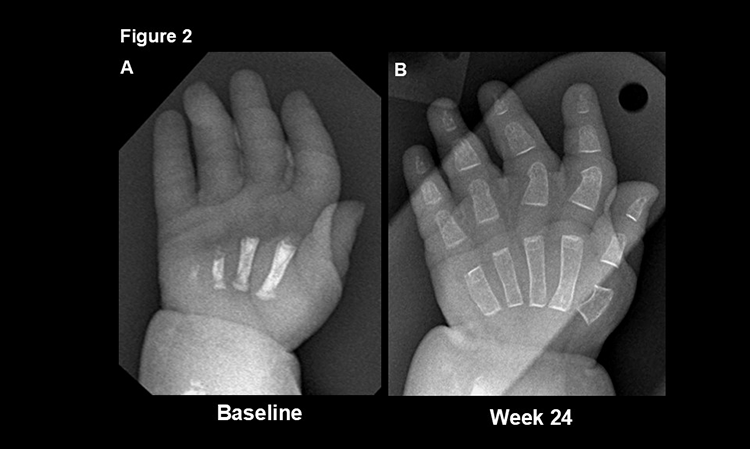
Hypophosphatasia (HPP) is an inborn error of metabolism, an inherited disorder in which mutations in the ALPL gene, which oversees how bones and teeth mineralize, measurably impair this process.
The result goes beyond soft, weak and sometimes mishappen bones. HPP is characterized by a range of symptoms that can make diagnosis difficult: breathing difficulties, fever, increased cranial pressure and issues with weight gain and growth—and even perinatal death in the most severe cases.

The effects of HPP can become noticeable within the first six months of a baby’s life, but they also appear throughout childhood and adulthood. The earlier HPP symptoms appear, the more severe the disease.
Milder cases put affected adults at greater risk of breaking bones. Severe forms cause life-threatening disease in approximately one per 100,000 live births.
There is no cure for HPP, but there is a dramatically mitigating treatment and the promise of improved diagnostics to come, in large part due to the work of José Luis Millán, PhD, a professor in the Center for Cardiovascular and Muscular Diseases.
Millán was among the first staff to join Sanford Burnham Prebys Medical Discovery Institute (then called the La Jolla Cancer Research Foundation) in 1977. Over the decades, he has focused much of his research on soft bone diseases, particularly HPP. He performed key research leading to the development and approval of Strensiq in 2015, a novel therapy that replaces a crucial but missing enzyme to boost bone mineralization.
“It’s been a tremendous success and has proven to be a lifesaving treatment,” said Millán. “Many children who have been treated otherwise would have died shortly after birth, and they are now able to look forward to long lives.”
Strensiq requires regular injections, however, prompting some patients to discontinue treatment. Millán and colleagues are now working on testing alternative treatments for HPP, such as a gene therapy that would effectively address HPP with a single treatment.
In a paper published in January 2025, they describe early efforts to deliver a gene (via a viral vector) that is capable of producing the missing enzyme for life. After a single injection, Mouse studies have produced encouraging results; the next step will be human clinical trials. “We have essentially titrated the viral vector to show which dose achieves efficacy without causing side effects, such as accumulations of bony crystals in soft organs called ectopic calcifications,” said Millán. “Our data provide a clear starting point for clinical trials.”
In a paper published in November 2025, The Millán laboratory also showed that oral administration of a small molecule drug can improve skeletal mineralization in mice displaying the later-onset form of HPP. “This treatment can fill a major clinical need for patients with the later-onset forms of HPP where enzyme replacement therapy is not approved.”